Tutorial 1. Basic DNA sequence manipulation¶
© 2018 Manuel Razo. This work is licensed under a Creative Commons Attribution License CC-BY 4.0. All code contained herein is licensed under an MIT license
Consider the Leviathan's teeth.¶
As we saw in lecture during the WoW (Week of Whales), the evolution of Cetaceans has served as an incredible puzzle for evolutionary biologists starting from Darwin himself. The transition from limbs-to-fins is one of the best documented evolutionary events within the fossil record. But in the age of modern DNA sequencing the cross-talk between classic paleontology and DNA science is still a rare event.
In the beautiful paper by Meredith et al. they exploit this interaction between the fossil record and the molecular evidence to "provide manifest evidence for the predictive power of Darwin's theory." In this work they are interested in the conservation of teeth among placental mammals. In particular they study the enamelin (ENAM) gene as a 'molecular fossil' that they can compare directly with morphological features well preserved in the fossil record.
The ENAM gene participates in the production and secretion of enamel, the hardest substance found in vertebrates that forms the outer cap of teeth. Being such a hard compound gives it a rich representation in the fossil record of placental mammals. But there are groups of toothless mammals (edentulous) such as pangolins, baleen whales, and anteaters, and mammals with enamelless teeth such as sloths, armadillos, sperm whales, and aardvarks.
If all of these "outlier" groups of mammals we know descended from a common ancestor that had enamel coated teeth since it is present in other vertebrate groups, Darwin's theory makes the simple prediction:
If the ENAM gene is present in the genome of these species it is likely to be pseudogenized.
What this means is that if one were to sequence the ENAM gene of edentulous and enamelless mammals and compare it with other placental mammals, there should be features such as stop codons, insertions or frame-shifts that would eliminate the functionality of the gene.
In this tutorial we will analyze the original data from the Meredith et al. paper in order to explore this pseudogenization hypothesis that they put forward. By doing so we will learn some basic commands to manipulate sequences in Python. The purpose of this and the following computational tutorials is to give you an idea of how powerful it is for evolutionary biologists to know how to write code.
We will begin by importing the necessary package to read the sequences. Usually we import all of our packages at the beginning of the file, but since this is the first tutorial we will import the packages as they are needed.
# Import bioinformatic tools
import Bio.AlignIO # To read sequence alignments
import Bio.Seq # Tools to manipulate sequences
Now that we have the needed package, let's read the file. We need to give the path to the directory where you have placed your data file. This will depend on how you have structured your folders. In my case I have created a folder called data directly in the folder where this notebook lives where I have placed the data file (called enamel_alignment.nxs).
Note: A .nxs file is a popular format that some phylogeny software use to save multiple sequence alignments.
# Define data directory
datadir = './data/'
# Read alignment file. For this we give a string that points at where the file
# is and then we indicate the format of the alignment. In our case Meredith et al
# save the alignment in the so-called nexus format.
enamel_aln = Bio.AlignIO.read(datadir + 'enamel_alignment.nxs', 'nexus')
# Print number of sequences in alignment
print('There are {:d} sequences in the alignment'.format(len(enamel_aln)))
A little bit about the syntax:
- The BioPython package which we import as
Biohas different modules within the package. What this means is that within the BioPython package the functions are organized into subgroups of specialized functions to perform certain tasks. For example here we use the moduleAlignIOwhich is a collection of routines to manipulate multiple alignment objects. So in order to read a multiple sequence alignment tool we must use thereadfunction from thisAlignIOmodule. - When we put
{:d}this serves as a placeholder for what will come after. Indicating admeans that it is a placeholder for anint, i.e. a variable that contains an integer number. The function.formatthat comes after the string is a method associated with variables of type string in which it will substitute the placeholder with whichever text we feed into the function.
We know that the alignment object has 49 entries. We showed that by using the function len. Let's now look at a single entry. Remember that Python starts indexing at zero, so in order to obtain the first entry we must type
enamel_aln[0]
We can see that each object is what is called a SeqRecord that contains different entries such as the sequence seq, the id of the sequence, the name and a description.
In order to access to each of these attributes we can simply type .attribute after the SeqRecord object and that will return whatever we want. For example if we want to extract the seq attribute from this first entry we can type
enamel_aln[0].seq
It would be useful to extract all of the sequences and all of the species names and save them in separate variables. For this we can use a very special type of for loop in Python.
Python has what is called list comprehensions are, in simple terms, one-liner for loops. For example let's say I want to loop from 0 to number 15 and I want to save the square of this number in an array. I could then just do the following:
my_array = [x**2 for x in range(16)]
my_array
Let's dissect this syntax. There are three components to a list comprehension:
- The function that will perform something with the variable we are looping through. In our case since we want to square the variable we indicate
x**2. - The variable itself. In our case
x. - The values of the variable that will be looped through. For our example since we wanted to loop from 0 to 15 we use the function
range(16)that returns an array containing the integer numbers from 0 to 15.
Now using list comprehensions let's extract into different lists both the sequences and the names of the species.
# Extract sequences and names for each entry in alignment
enamel_seq = [record.seq for record in enamel_aln]
enamel_name = [record.name for record in enamel_aln]
Now that we extracted the sequences we can start investigating the hypothesis put forward by the authors. The claim is that they found that in all 3 open reading frames (ORFs) there were stop codons only in the edentulous and the enamelless species. So in order to find stop codons we need to take these DNA sequences and translate them into amino-acids. Each of our sequences, not being regular Python strings, but rather BioPython sequence objects have a translation method associated with them. Let's try it in one of our sequences.
enamel_seq[0].translate()
What happened? The error says that
TranslationError: Codon '?GA' is invalid
Since these sequences come from a multiple alignment many of the characters are either ? if there was no basepair to be aligned at that possition (for example if a sequence started downstream from the others or ended before), or - if there was a gap in the alignment. That means that keeping the sequences as they are won't allow us to test our hypothesis. We need to remove these characters.
If one googles for ≈ 30 seconds we can find that sequence objects have another method ungap that removes any type of character, but only one at the time. So if we want to remove both characters we need to do it in two steps. Let's first show it for a single sequence.
# Extract single sequence
seq = enamel_seq[0]
# Remove the missing "?" character
seq = seq.ungap('?')
seq
Now let's remove the second character
# Remove the gap character "-"
seq = seq.ungap('-')
seq
In principle we should be able to translate this sequence, so let's test it!
seq.translate()
It worked! Biopython is giving us a warning message that our sequence is not a multiple of three and suggest that we trim the sequence because in the future this could be an error. We can easily do that by figuring out the residual of dividing the length of the sequence by three. That is len(seq) modulo 3.
# finding number of letters to trim
trim_char = len(seq) % 3
# trim sequence to translate it
trim_seq = seq[:-trim_char]
trim_seq
The syntax that we used [:-trim_char] means that Python should take all the characters up to the trim_char counting backwards. Try defining a list yourself with multiple objects and index it using [-1]. You'll see that it will return the last element of the list. On the other hand if you index it using [:-1] it will return all elements except the last one.
Note that we trim from the right-hand side of the equation because if we were to trim from the left we would actually be chaning the way that the codons are read.
Now we should be able to translate this sequence without the warning.
trim_seq.translate()
Excellent! Let's now clean all the sequences again using the powerful list comprehensions.
# Let's remove the missing "?" and gap "-" characters from all records
enamel_clean_seq = [seq.ungap('?') for seq in enamel_seq]
enamel_clean_seq = [seq.ungap('-') for seq in enamel_clean_seq]
The function translate just translates one of the ORFs. But we need to translate all three ORFs in order to test the hypothesis that Meredith et al. presented.
What we are going to do is we are going to build a lookup table with the following columns:
- name : To save the name of the species
- frame : To save the number of ORF
- DNA : To save the associated DNA sequence
- protein : To save the translation of the DNA sequence for the given frame
This means that each of the species will have 3 entries in the table, one per ORF. For this we will use the DataFrame structure from the powerful pandas package. Over the next tutorials we might make more and more use of this powerful package since it is one of the cornerstones of the Python environment that allows us to manipulate data very easily.
First let's import the package. Again normally this will go in the very first cell of the notebook, but since we are just learning let's import it here.
import pandas as pd # For data manipulation
Now we will initialize an empty data frame that already has the columns that we indicated we want.
# Initialize DataFrame to save translations for each ORF.
columns = ['name', 'frame', 'DNA', 'protein']
df_prot = pd.DataFrame(columns=columns)
df_prot
The next thing we need to do is loop through each of the sequences, and then loop through each of the 3 ORFs translating at each iteration of the sequence. Let's list the steps that we need to follow then:
- Extract the species name.
- Find the length of the sequence given the frame
- Extract the corresponding DNA sequence by trimming it from both sides, on the left side to choose the current ORF, and on the right side to make it a multiple of three.
- Translate the DNA sequence into amino-acids.
- Convert all the relevant entries into a
pandas Series. This step is necessary because if we want to add a new row to ourDataFrameit has to be via aSeriesobject. - Append the entry into the DataFrame.
Let's then implement this!
# Loop through each of the sequences
for i, seq in enumerate(enamel_clean_seq):
# 1. Extract the species name
species_name = enamel_name[i]
# Loop through each of the available ORFs
for frame in range(3):
# 2. Find the length of the sequence given the current ORF
# For this we will take the sequence from position frame
# (either 0, 1 or 2) all the way to the end. That is indicated as
# [frame:]. Then we will subtract the length modulo 3 to trim the
# extra residues.
orf_len = len(seq[frame:]) - len(seq[frame:]) % 3
# 3. Extract the corresponding DNA sequence.
orf_seq = seq[frame:frame+orf_len]
# 4. Translate the DNA sequence
orf_prot = orf_seq.translate()
# 5. Convert into pandas Series.
# Series don't have column names, but they have "index" names. So
# we will assign the same names to these indexes as the column names
# of our DataFrame. We will also convert the sequences to strings since
# Pandas saves Bio sequences in a weird format
orf_series = pd.Series([species_name, frame, str(orf_seq), str(orf_prot)],
index=columns)
# 6. Append entry to the DataFrame
df_prot = df_prot.append(orf_series, ignore_index=True)
# Print the head of the table
df_prot.head()
For the for loop we used the convenient enumerate function. What this does is that it assigns an index to each of the objects we loop through. In our case we indicated for i, seq in enumerate(enamel_clean_seq). This means that the variable seq will contain the actual sequence object, and the variable i will have the index (0, 1, 2,$\ldots$) that enumerates each of the sequences.
Once we have all of the protein sequences we need to count the number of stop codons per sequence. These codons are indicated by * symbols. So it is just a matter of counting the number of these characteres per entry in our data frame. For this we can use the method .count() associated with strings. Let's quickly look at an example.
# Define a simple sequence
string = 'hello, world!'
# Count the numer of `l` characters
print('number of l in the string: ', string.count('l'))
print('number of o in the string: ', string.count('o'))
It is important to highlight that the sequences used in this exercise covered ≈ 80% of the length of the ENAM gene. That is why we don't worry about the location of the stop codons since if present they would at least remove 20% of the gene, making it most likely useless.
Now let's loop through each of the entries in our data frame, saving at each iteration the number of * characters. Let's then append these entries as an extra column to our DataFrame.
# Initialize list to save open reading frames
stop_codons = []
# Loop through protein sequences counting stop codons
for prot in df_prot.protein:
stop_codons.append(prot.count('*'))
# Append number of stop codons as column to the dataframe
df_prot['num_stops'] = stop_codons
# Print head of expanded DataFrame
df_prot[['name', 'frame', 'num_stops']].head()
Now that we have the number of stop codons per sequence we need to find which species have at least one stop codon in all 3 ORFs. One of the powerful features about a pandas DataFrame is the way we can access the data. For example let's say we are interested in all the entries that have the aardvark species Orycteropus afer. For this we can use boolean indexing.
Boolean indexing allows us to feed an array full of True and False statements as an index for our DataFrame and it will only return the rows that were true. Let's look at this example.
df_prot[df_prot['name'] == 'Orycteropus_afer']
This is super awesome! We can use this to extract species by species the number of stop codons and then ask if all of them are greater than zero or not. This should return True if each ORF had at least one stop codon and False otherwise.
Let's look at how this would work. Using the same example as above let's ask if all the num_stops entries from the aardvark are greater than zero.
df_prot[df_prot['name'] == 'Orycteropus_afer'].num_stops > 0
As it is this returns the individual boolean statements. If we use the method .all() this will return a single boolean variable. It will be True if and only if all entries are True, and False otherwise.
(df_prot[df_prot['name'] == 'Orycteropus_afer'].num_stops > 0).all()
Let's now generate a new DataFrame where we save the name of the species and a column named all_stops that contains a boolean variable indicating if all ORFs had at least one stop codon.
# Initialize a DataFrame to save per species whether or not all
# ORFs have stop codons
df_stops = pd.DataFrame(columns=['name', 'all_stops'])
# Loop through species and ask if all frames have a stop codon
for species in enamel_name:
# Extract the number of stops for the current species
stops = df_prot[df_prot.name == species].num_stops
# Ask if all ORFs have stop codons
species_stop = (stops > 0).all()
# Append result to DataFrame
df_stops = df_stops.append(pd.Series([species, species_stop],
index=['name', 'all_stops']),
ignore_index=True)
# Print header of DataFrame
df_stops.head()
Eureka! We now have the information we need to test Darwin's hypothesis. Let's find which species had a stop codon in all three ORFs. We can again use the powerful concept of boolean indexing asking which rows have a True in the all_stops column.
df_stops[df_stops.all_stops == True].name
Let's compare this list to the result from the Meredith et al. paper.
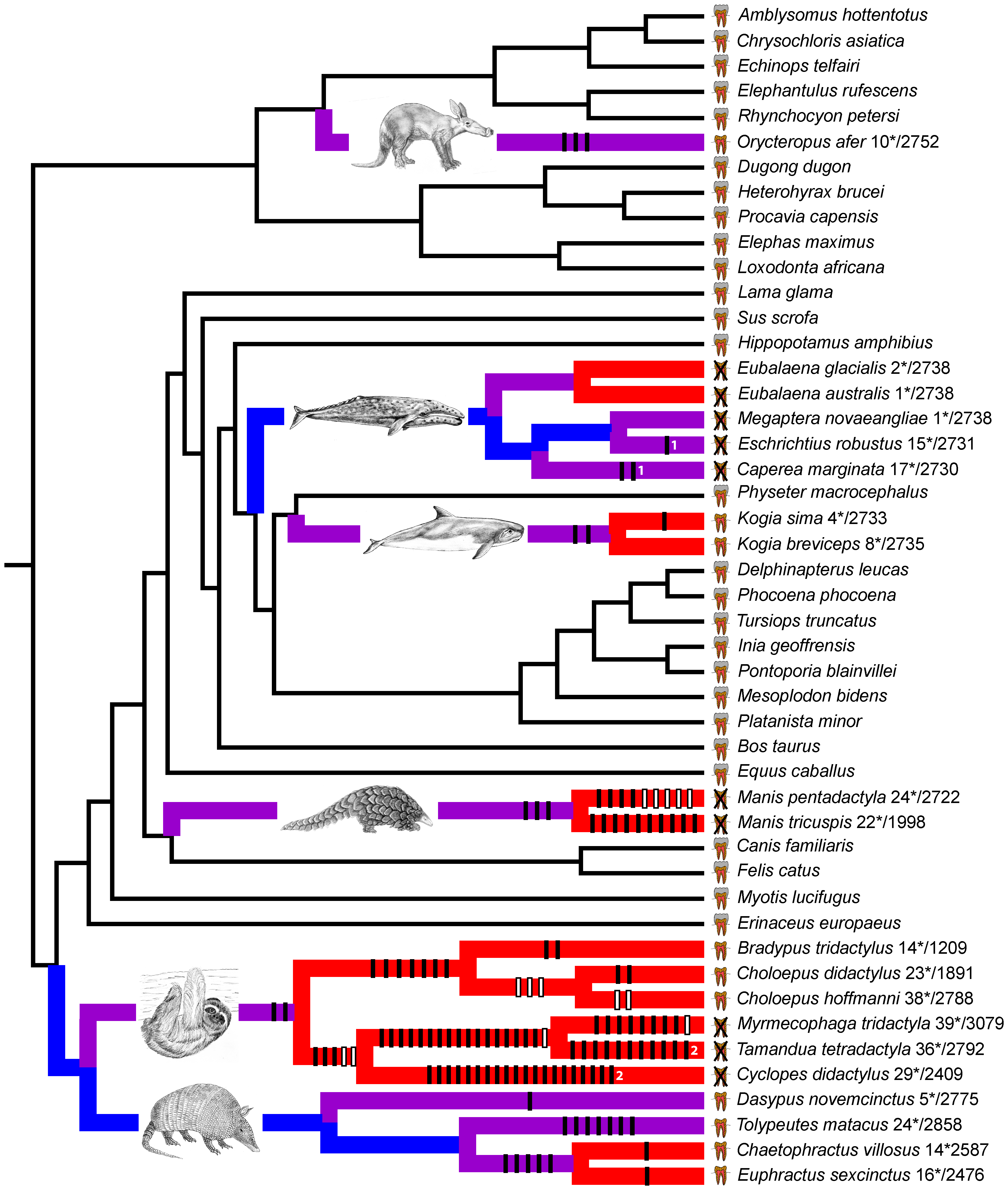
A close inspection of this tree will reveal that the 20 species that we found to have stop codons in all ORFs are all of the species in this tree that either have no teeth or their teeth contain no enamel! So yet another proof of the predictive power of Darwin's theory.
What we've learned¶
In this tutorial we learned how to perform basic sequence manipulations such as removing gaps, indexing and translating from DNA to amino-acids.
We also learned about the power of using pandas to contain and manipulate our data.
All of this with the final goal of testing a prediction that falls directly from Darwin's theory and our modern understanding of genetics. This hopefully gave you an idea of how powerful coding can be in the modern era of DNA sequencing.